



.svg)
.svg)

Die Richtlinien der Europäischen Union (EU) zur Guten Herstellungspraxis (GMP) und zur Guten Vertriebspraxis (GDP) sowie das Europäische Arzneibuch (Pharmacopoeia Europaea, Ph. Eur.) und das Schweizerischen Pharmakopöe (Pharmacopoea Helvetica, Ph. Helv.) sind verbindlich für Marktteilnehmer, die gewerblich mit Arzneimitteln und Medizinprodukten umgehen. Die Rechtsordnung der EU steht in diesem Zusammenhang über der Rechtsordnung der EU-Mitgliedsstaaten und umfasst Verordnungen und Richtlinien.
Wichtig ist dabei, die Kontrollinstanzen, die geltenden Normvorschriften, wichtige Begriffe und die Fristen für die Kalibrierung zu kennen.
Kontrollorgane
Deutschland
In Deutschland führen Bundesbehörden genehmigungsbezogene Kontrollen durch. Darüber hinaus ist in der Regel die zuständige Behörde des jeweiligen Bundeslandes vor Ort zuständig, d.h. Landesbehörden, Bezirksverwaltungen, Landesdirektionen oder Staatsministerien in den jeweiligen Bundesländern. Sie führen Kontrollen durch und werden tätig, falls sie Verstösse gegen die Vorschriften feststellt.
Darüber hinaus sind die Behörden für den Gesundheitsschutz zuständig (u.a. die Überwachung durch Inspektionen nach GMP, GDP, etc.). Je nach Bundesland sind die Zuständigkeiten unterschiedlich geregelt. Welche Behörde in welchem Bundesland welche Aufgaben wahrnimmt, können Sie dem Verzeichnis der zuständigen Behörden, Ämter und Sachverständigen entnehmen, die für die Durchsetzung des deutschen Arzneimittelgesetzes (AMG) zuständig sind.
Eine wichtige koordinierende Funktion hat die Zentralstelle der Länder für Gesundheitsschutz bei Arzneimitteln und Medizinprodukten (ZLG) mit Sitz in Bonn. Die ZLG sorgt für eine durchgängige Überwachung, koordiniert länderübergreifende Massnahmen und Inspektionen im zentralen Zulassungsverfahren und tauscht Informationen mit europäischen und internationalen Überwachungsstellen aus. Ausserdem koordiniert sie die Aktivitäten der Arzneimitteluntersuchungsstellen in den Bundesländern.
Schweiz
Swissmedic ist die zentrale Schweizerische Zulassungs- und Aufsichtsbehörde für Arzneimittel und Medizinprodukte. Als öffentlich-rechtliche Anstalt des Bundes mit Sitz in Bern ist sie in ihrer Organisation und Führung unabhängig und verfügt über ein eigenes Budget. Swissmedic selbst ist für die Kontrolle von Blut und seltenen Prozeduren zuständig und verantwortet Zulassungen und Betriebsbewilligungen.
Kontrolle von Heilmitten:
Die kantonalen Inspektorate sind für die allgemeinen GMP/GDP-Kontrollen zuständig. Sie werden von Swissmedic beaufsichtigt und koordiniert. In der Schweiz gibt es die folgenden Inspektorate:
- Regionales Heilmittelinspektorat der Nordwestschweiz (RHI NW)
- Regionale Fachstelle der Ost- und Zentralschweiz (RFS-OZ)
- Regionales Medizin-Inspektorat der Südschweiz, IRM-S
U.S. Food and Drug Administration (FDA)
Die Food and Drug Administration (FDA) ist die Aufsichtsbehörde der Vereinigten Staaten von Amerika für Lebensmittel und Arzneimittel. Die FDA und andere ausländische Behörden können von lokalen Behörden (z. B. der Swissmedic) ermächtigt werden, Schweizer Unternehmen zu überprüfen.
Normen und Vorschriften
GxP
GxP ist die Abkürzung für die allgemeine gute Praxis, die sich auf eine Reihe von Gesetzen, Bestimmungen und Leitlinien bezieht, die verschiedene Bereiche der Forschung, Entwicklung, Prüfung, Herstellung und des Vertriebs von Arzneimitteln regeln. Das "x" kann durch die im Folgenden erläuterten Buchstaben ersetzt werden.
GLP (Good Laboratory Practice)
Unter guter Laborpraxis (GLP) versteht man Richtlinien zur Qualitätssicherung von Rahmenbedingungen und organisatorischen Abläufe, unter denen nicht-klinische Gesundheits- und Umweltsicherheitsprüfungen geplant, durchgeführt und überwacht werden. Dazu gehören auch die Aufzeichnung, Archivierung und Berichterstattung der Prüfungen.
GMP (Good Manufacturing Practice)
Die gute Herstellungspraxis (GMP) ist ein Leitfaden für die Qualitätssicherung der Produktionsumgebung und der Produktionsprozesse bei der Herstellung von Arzneimitteln und pharmazeutischen Wirkstoffen sowie von Kosmetika, Lebens- und Futtermitteln.
GDP (Good Distribution Practice)
Die gute Vertriebspraxis bezieht sich auf Leitlinien für die Qualitätssicherung in der gesamten Lieferkette von pharmazeutischen Wirkstoffen und Arzneimitteln.
Pharmacopoeia Europaea (Ph. Eur.)
Das Arzneibuch wird von der EU als European Pharmacopoeia herausgegeben und enthält risikogerechte, anerkannte Qualitätsvorschriften für gängige und bekannte Arzneimittel und pharmazeutische Hilfsstoffe. Die darin enthaltenen Bestimmungen sind verbindlich und haben Gesetzeskraft. Die Pharmacopoeia trägt damit wesentlich dazu bei, dass Heilmittel für alle Patienten in gleichbleibend hoher Qualität zur Verfügung stehen und schafft damit eine zentrale Voraussetzung für sichere und wirksame Heilmittel.
Pharmacopoea Helvetica (Ph. Helv.)
Das Schweizerische Arzneibuch besteht aus der European Pharmacopoeia und der Schweizerischen Pharmakopöe (Pharmacopoea Helvetica - Ph. Helv.). Es wurde von Swissmedic auf der Grundlage des Schweizerischen Heilmittelgesetzes (HMG) herausgegeben. Es enthält risikogerechte, anerkannte Qualitätsanforderungen für gängige und bekannte Arzneimittel, pharmazeutische Hilfsstoffe und einzelne Medizinprodukte.
ISO/IEC 17025:2017
Diese internationale Norm legt die allgemeinen Anforderungen an die Kompetenz bei der Durchführung von Prüfungen und/oder Kalibrierungen, einschliesslich der Entnahme von Proben, fest. Eine Akkreditierung nach ISO 17025 bedeutet, dass die Prüf- und/oder Kalibrierlabore über technische Kompetenz verfügen und in der Lage sind, fachlich fundierte Ergebnisse zu liefern. Akkreditierungen nach ISO 17025 werden in der Schweiz zentral von der Schweizerischen Akkreditierungsstelle SAS durchgeführt.
ISO 13485
Die ISO-Norm 13485 stellt die Anforderungen an ein umfassendes Qualitätsmanagementsystem für die Entwicklung und Herstellung von Medizinprodukten dar. Die Norm enthält detaillierte Anforderungen zu Themen rund um das Design, die Herstellung und das Inverkehrbringen von Medizinprodukten. Die Einhaltung der Norm wird durch Akkreditierung nachgewiesen und kontinuierlich überwacht.
ISPE GAMP 5
Der Good Automated Manufacturing Practice (GAMP) Leitfaden für die Validierung von automatisierten Systemen in der pharmazeutischen Produktion wurde in Zusammenarbeit mit der International Society for Pharmaceutical Engineering (ISPE) veröffentlicht. Der GAMP-Leitfaden ist als Richtlinie ohne rechtlich bindenden Charakter gedacht. Dennoch ist er zum Standardregelwerk für die Validierung computergestützter Systeme (CSV) von Herstellern und Zulieferern in der pharmazeutischen Industrie geworden. Auf der Website der ISPE heisst es zu GAMP 5:
"GAMP 5: Ein risikobasierter Ansatz für GxP-konforme computergestützte Systeme bietet eine pragmatische und praktische Anleitung für die Industrie, mit dem Ziel, auf effiziente und effektive Weise regelkonforme computergestützte Systeme zu schaffen, die für den beabsichtigten Gebrauch geeignet sind und gleichzeitig Innovation und technologischen Fortschritt ermöglichen." [URL: https://ispe.org/product-types/gamp-good-practice-guides].
FDA 21 CFR Part 11
FDA 21 Code of Federal Regulation, Part 11 (FDA 21 CFR Part 11) ist ein Gesetz der Vereinigten Staaten von Amerika. Darin formuliert die FDA Anforderungen an elektronische Aufzeichnungen und Signaturen, die sich an Hersteller von Arzneimitteln und medizinischen Geräten richten. FDA 21 CFR Part 11 gilt immer dann, wenn Informationen elektronisch erstellt, geändert, gespeichert, übertragen oder abgerufen werden sollen.
IATA
Die International Air Transport Association (IATA) wurde als Dachverband der Fluggesellschaften gegründet. Ziel der IATA ist es unter anderem, den sicheren, planmässigen und wirtschaftlichen Transport von Personen und Gütern auf dem Luftweg zu fördern – was implizit auch Arzneimittel und Medizinprodukte einschliesst.
Wichtige Begriffe
Validierung
Die Validierung erbringt den dokumentierten Nachweis, dass ein Prozess oder System die zuvor festgelegten Anforderungen (Akzeptanzkriterien) im praktischen Einsatz reproduzierbar erfüllt. Voraussetzung für eine Qualifizierung ist in der Regel eine Validierung des dazugehörigen Prozesses. Um beispielsweise einen Transportprozess zu validieren, müssen auch die im Prozess eingesetzten Fahrzeuge, Temperatursensoren, Mitarbeiter etc. entsprechend qualifiziert werden.
Qualifizierung
Bei der Qualifizierung von Fachkräften, Geräten und Einrichtungen wird deren Eignung für die vorgesehene Aufgabe mit der eingesetzten Technik überprüft. Eine Qualifizierung ist also der dokumentierte Nachweis, dass ein Raum, ein System, eine Anlage, ein Lieferant oder ein Mitarbeiter allein oder im Zusammenwirken für den vorgesehenen Zweck geeignet ist. Werden auch physikalische Grössen gemessen, ist die Kalibrierung eine Voraussetzung für die Qualifizierung.
Unterschied zwischen Validierung und Qualifizierung
Für die Unterscheidung zwischen Validierung und Qualifizierung gibt es eine grobe Faustregel: Was Sie anfassen können, ist qualifiziert, was Sie nicht anfassen können, ist validiert.
Akkreditierung
Ein Prüf- oder Kalibrierlabor ist akkreditiert, wenn es beispielsweise die Anforderungen der Norm DIN EN ISO/IEC 17025 erfüllt. Dazu wird das Labor von Gutachtern einer unabhängigen Akkreditierungsstelle, die der Norm DIN EN ISO/IEC 17011 entspricht, beurteilt und in Abständen von ein bis eineinhalb Jahren überprüft.
Zertifizierung
Die Zertifizierung ist ein Verfahren, mit dem die Einhaltung bestimmter Anforderungen nachgewiesen werden kann. Zertifizierungen sind ein Teilprozess der Konformitätsbewertung. Sie werden häufig von unabhängigen Zertifizierungsstellen für einen begrenzten Zeitraum ausgestellt. Ein Beispiel ist die Ausstellung eines auf ein Jahr begrenzten Zertifikats für einen Temperatursensor.
Kalibrierung
Kalibrierung in der Messtechnik ist ein Messprozess zur zuverlässigen und reproduzierbaren Ermittlung und Dokumentation der Abweichung eines Messgerätes oder einer Massverkörperung gegenüber einem anderen Gerät oder einer anderen Massverkörperung, die in diesem Fall als normal bezeichnet werden.
Justierung
Bei der Justierung wird ein Gerät technisch verändert, um eine möglichst geringe Abweichung, zum Beispiel des Temperatursensors, zu erreichen. Nach dem Abgleich muss stets eine Kalibrierung durchgeführt werden.
Wichtige Abkürzungen
Viele Fachleute im Bereich Pharmazeutika und Medizinprodukte verwenden Abkürzungen, die Prozesse, Bestimmungen und Instanzen bezeichnen. Eine Auswahl häufig verwendeter Abkürzungen finden Sie in der nachstehenden Tabelle:
| Abkürzung | Englischer Begriff | Deutscher Begriff |
| API | Active pharmaceutical ingredient | Wirkstoff |
| CAPA | Corrective and preventive action | Korrektur- und Vorbeugemassnahme |
| FDA | Food and Drug Administration | U.S. Lebensmittel- und Arzneimittelbehörde |
| GAMP | Good automated manufacturing practice | Gute automatisierte Fertigungspraxis |
| GCP | Good clinical practice | Gute klinische Praxis |
| GDP | Good distribution practice | Gute Vertriebspraxis |
| GLP | Good laboratory practice | Gute Laborpraxis |
| GMP | Good manufacturing practice | Gute Herstellungspraxis |
| IATA | International Air Transport Association | Internationaler Luftfahrtverband |
| IMP | Investigational medicinal products | Arzneimittel für eine klinische Studie |
| ISO | International Organization for Standardization | Internationale Organisation für Normung |
| ISPE | International Society for Pharmaceutical Engineering | Internationale Gesellschaft für Pharmazeutische Technik |
| PIC | Pharmaceutical Inspection Convention | Pharmazeutische Inspektionskonvention |
| PiT | Product in transit | Produkt auf dem Transportweg |
| SAS | Swiss Accreditation Service | Schweizerische Akkreditierungsstelle |
| SCS | Swiss Calibration Service | Schweizerischer Kalibrierdienst |
| SW | Software | Software |
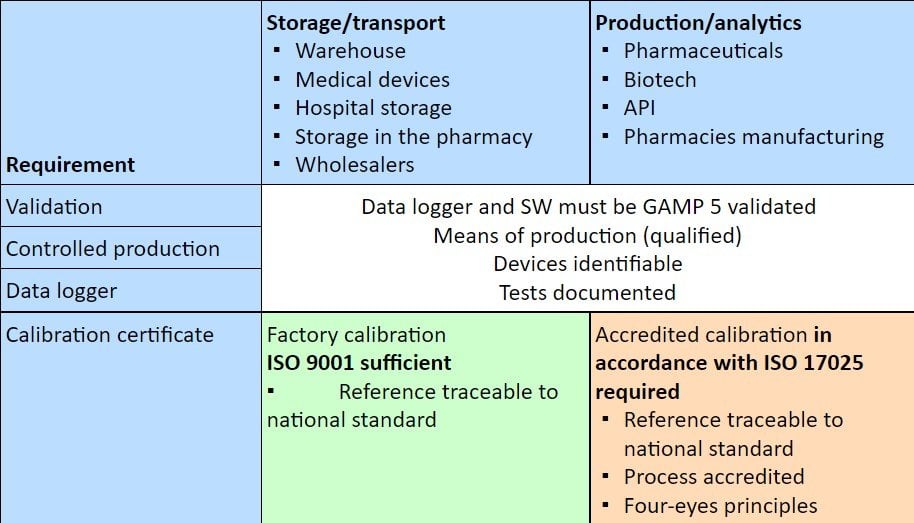
Wann ist welche Kalibrierung erforderlich?
Die folgende Tabelle zeigt, welche Art der Kalibrierung in welchem Bereich der pharmazeutischen Lieferkette von den Behörden gefordert oder erwartet wird.
Fazit
Der Anforderungskatalog für den Umgang mit pharmazeutischen Produkten und Medizinprodukten innerhalb der gesamten Lieferkette ist vielfältig und mag manchem wie ein Normen-Dschungel erscheinen. Die Gefahr, etwas zu übersehen oder zu vergessen, ist allgegenwärtig. Gerade im Hinblick auf eine ausreichende Kalibrierung stellt sich immer mehr die Frage, was die Behörden zu Recht fordern und was eigentlich eine Übererfüllung darstellt. Selbstverständlich sind die Anforderungen in den verschiedenen Gesetzen, Richtlinien, Verordnungen usw. verbindlich und damit zwingend vorgeschrieben.
